A.D. Walsh, Nature 1947, 159, 712-3
copyright © 1947 Macmillan Magazines Ltd.
Structures of Ethylene Oxide and Cyclopropane
MY suggestion1 that ethylene oxide and cyclopropane might be portrayed as (I)-(II) rather than (III)-(IV) has been criticized.2,3 I wish, therefore, to discuss my suggestion further.
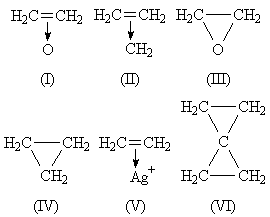
My underlying ideas were: (1) evidence on olefinic donor properties suggests (I)-(II), just as (V) is known; (2) the carbon atoms involved are nearer the trigonal or ethylenic than tetrahedral state. Evidence for (2) seems very strong. (a) It is outstanding that epoxy or cyclopropyl groups joined to C=C show just those spectroscopic and chemical properties characterizing conjugation. Theory explains conjugation thus. All the carbon atoms involved are in the trigonal state: 2p atomic orbitals on neighbouring carbon atoms overlap to give a nonlocalized molecular orbital. The conjugation phenomena follow from the delocalization. Hence the idea that ethylene oxide and cyclopropane contain near-trigonal carbon atoms and molecular orbitals which, being built from 2p atomic orbitals, inevitably overlap with neighbouring 2p atomic orbitals. (b) The low dipole moment of cyclopropyl chloride: the high electronegativity of a trigonal carbon atom in its hybrid valencies,5 as in vinyl chloride, explains this. (c) The height of the strong CH Raman frequencies and the unsaturation properties of ethylene oxide and cyclopropane.
One is therefore led to structures with carbon atoms near the trigonal state. (I)-(II) suggest the existence of trigonal carbon atoms, non-localized orbitals, unsaturation, conjugating power. (III)-(IV) do not.
(I) suggests resemblance to amine oxides. Such is seen in work by Karrer,4 to which Sir Robert Robinson does not refer: certain epoxy groups do lose oxygen readily (→ C=C). I do not suppose likeness between (I) and amine oxides to be very close: the orbital symmetries involved are so different. Similarly, analogy between (I) and (V) is not very close, for O, unlike Ag+, has further valency orbitals which must be involved: their importance can be seen below.
It is clear that the probable symmetry (from infrared data: not from intuition, which would suggest O3 to be equilateral) of cyclopropane enforces superposition of three forms like (II). The resultant is not (IV), for it is still based on trigonal rather than tetrahedral carbon atoms. In the hybrid, the CH2 planes must bisect the CCC angles: molecular orbitals are formed by overlap of (a) three hybrid sp2 atomic orbitals, (b) 2p atomic orbitals. (VII) shows this: AB, CB, DB are CH2 planes at 90° to the paper. The result has some of the character of (VIII). Two electrons occupy the three-pronged area a, two may be found either in b or d or e, and two lie in an orbital that represents a first overtone of a. The latter are 'anti-bonding' electrons: their presence reflects repulsion in (II) between the lone pair and other electrons. b, d, e are regions of high amplitude in an orbital with no actual nodes.
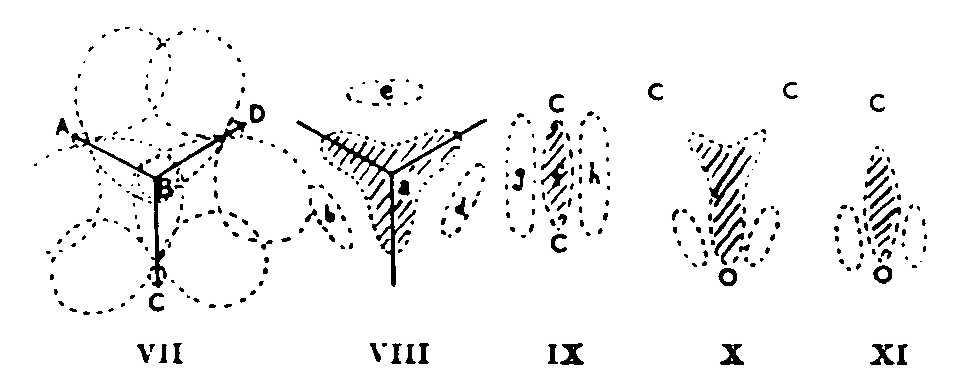
The resonance description of cyclopropane on my model is as a hybrid of three forms (II), the molecular orbital description partly as (VIII). The orbital pattern (VIII) to which we have been led is a generalization to three centres of the orbital picture (IX), which theory has evolved to explain two-centre unsaturation and make redundant the old distorted tetrahedral model for ethylene. f represents the s, g-h the p orbital. (That (IX) and (VIII) are the same geometrical pattern applied to two and three centres respectively can be shown by folding filter papers along (i) two diameters at 90°, (ii) three at 60°, making the same cuts in each and opening out; f becomes a, g-h become b-d-e.) The molecular orbital picture for ethylene oxide is as (VIII), except that the dipole means e is now tenuous, b-d drawn downwards, the upper prongs contracted. (X) ensues. One reaches the same essential result5 by considering the single structure (I) as the major resonance form. A C=O group has the molecular orbital picture (XI). Some resemblance of ethylene oxide to CH3CHO may therefore follow. Cationoid character comes from a large partial positive charge on the CC of (X), as on C of C=O.
(VII) implies, besides (VIII), some localized-bond character. The overlap between any pair of 2p atomic orbitals is neither 'sideways' nor 'endwise'. So far as it is the latter, the resulting molecular orbital overlaps little with the third 2p atomic orbital, that is, localized bonds at b, d, c result, a is vacated and its component atomic orbitals, mixed with the original carbon atomic orbitals, cause some change towards tetrahedral atoms. The angle between the 2p axes is such that sideways overlap, with resulting non-localization, is greater than endwise -- in accord with the carbon atoms being nearer trigonal than tetrahedral. In cyclobutane the angles are such that localization is greater and four-centre unsaturation less. Even in cyclohexane, however, a greater tendency (than in paraflins) towards 2p character in the C-C and of 2s in the C-H valencies is shown by CC weakness and CH strength.5
In azoxy compounds, lone pairs and not bonding electrons are donated. This is not against (I), as ethylene has no lone pair. It is significant that azoxy groups do not form strained rings, though far less distortion of natural valency angles is necessary than in (I). A most important stable molecule to consider is spiropentane, usually drawn as (VI). My ideas are readily extended to this molecule, the central carbon atom being digonal and the two CCC planes at 90°.
Dr. McDowell's arguments seem not to affect my model. I do not expect (I)-(II) to show olefinic frequencies: the disturbance is too great for this. I expect S+-R to be able to replace NH or CH2 or O.
So far as I can see, my model explains qualitatively all the salient facts. I think, therefore, (I)-(II) possible single formulæ, full descriptions (especially with (II)) needing hybridization with like forms; but if entirely new symbols for the binding at issue be suggested, I shall quite agree. I have been mainly concerned to apply wave mechanical principles to the problem -- though Sir Robert Robinson apparently does not realize this. As regards co-ordination by bonding electrons, my contribution was to try to combine clear statement with a rational basis for assessing donation probability, namely, ionization potential knowledge.
A. D. WALSH
Laboratory of Physical Chemistry,
Cambridge.
Dec. 7.
- Walsh, Nature, 159, 165 (1947).
- Robinson, Nature, 159, 400 (1947).
- McDowell, Nature, 159, 508 (1947).
- Karrer, Helv. Chim. Acta, 28, 474 (1945).
- Walsh, in course of publication.