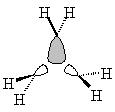
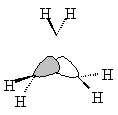
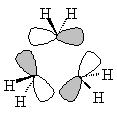
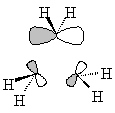
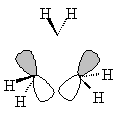
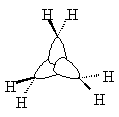
| Sketched orbitals | Calculated orbitals |
It should be qualitatively apparent that the a1' MO (ψA1') is more bonding than the MOs ψE',c and ψE',d; therefore we place ψA1' below the e' pair. And since the bonding combination of the methylene a1 orbitals is more bonding than the bonding combination of the methylene b2 orbitals, we also expect the antibonding combinations ψE',a and ψE',b to be higher in energy than the antibonding orbital ψA2'.
Extended Hückel calculations bear out this qualitative analysis.
| Orbitals | Energies | |||
| ψE',a | |
ψE',b | |
+6.8 eV |
| ψA2' | |
-3.4 eV | ||
| ψE',c | |
ψE',d | |
-13.2 eV |
| ψA1' | |
-15.4 eV | ||
| Derivation of the MOs | Walsh Cyclopropane home | Conclusions |
| Calculated orbitals | ||
Copyright © 1997 by Daniel J. Berger. This work may be copied without limit if its use is to be for non-profit educational purposes. Such copies may be by any method, present or future. The author requests only that this statement accompany all such copies. All rights to publication for profit are retained by the author.
Images generated with HyperChem Lite and ChemWindow Suite.